
Publications
Software as Medical Device: New European Commission Guidance
The European Commission’s Medical Devices Coordination Group has published much anticipated guidance on the qualification and classification of software under the new Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR). The guidance provides important clarification on which types of software will be regulated and how they should be classified and placed on the market.
The remit of when software will qualify as medical device software (MDSW, previously termed SMD or SaMD) has remained the same as under previous commission guidance, European Medical Device Vigilance System (MEDDEV) 2.1/6. The determining factor for qualification as MDSW remains whether the software has an “intended medical purpose.”
Undoubtedly the biggest change introduced by the new MDR is the up-classification of MDSW, which is clarified by the new guidance. Under the currently applicable rules, most stand-alone software is classified as Class I (low risk) devices. However, Rule 11 of the MDR significantly changes the classification of MDSW. The guidance interprets it as meaning that the default classification for all MDSW under Rule 11 is Class IIa (medium risk) unless there are reasons for a higher classification up to Class III (high risk), as all MDSW is software intended to assist decisions for diagnostic or therapeutic purposes. Likewise, software monitoring physiological processes will now be classified as Class IIa or higher. Most MDSW products currently classified as Class I are expected to be upgraded to Class IIa and above, thus requiring the involvement of a notified body (NB) to ensure that the MDSW can receive a CE marking.
The up-classification is illustrated by examples set out in the new guidance, which also cross refers to the frequently updated Borderline Manual (currently under revision for adaptation to the MDR). Some examples set out in the guidance reflect novel technologies and clarify that MDSW intended to perform diagnoses by analyzing images for treatment decisions, or software with a diagnostic function enabling a treatment option would bear a higher classification, depending on the effect of this treatment decision output.
Another change explained by the guidance is the importance of determining whether the software:
- is independent,
- is considered as an accessory to a hardware medical device, or
- “drives or influences” a medical device.
Software “driving or influencing the use of a (hardware) medical device” may be achieving its own intended purpose and will therefore be classified on its own. It will have at least the same risk class as the hardware medical device but possibly a higher one. This is in contrast with the current position of such software automatically falling into the same class as the device it drives. In the event of several rules applying to a device, the strictest rule resulting in higher classification will be followed.
The new guidance clarifies that these new rules are aimed at ensuring alignment of the classification rules in Europe with international standards, per the principles set out in the International Medical Device Regulators Forum (IMDRF) guidance. Therefore, a classification is assigned based on the importance and the impact of the information produced by the software within the healthcare decision process.
This has multiple consequences, including the fact that image analysis software might bear a classification higher than the device this software drives, and that a modular approach to qualification of MDSW might not be possible in instances where the function of technical (not per se medical) software affects the operation of the MDSW. For instance, image optimization software might not be able to escape CE-marking if the MDSW output is affected. This does not necessarily change the current position, but it makes it more explicit and suggests that a higher classification might apply to certain MDSW.
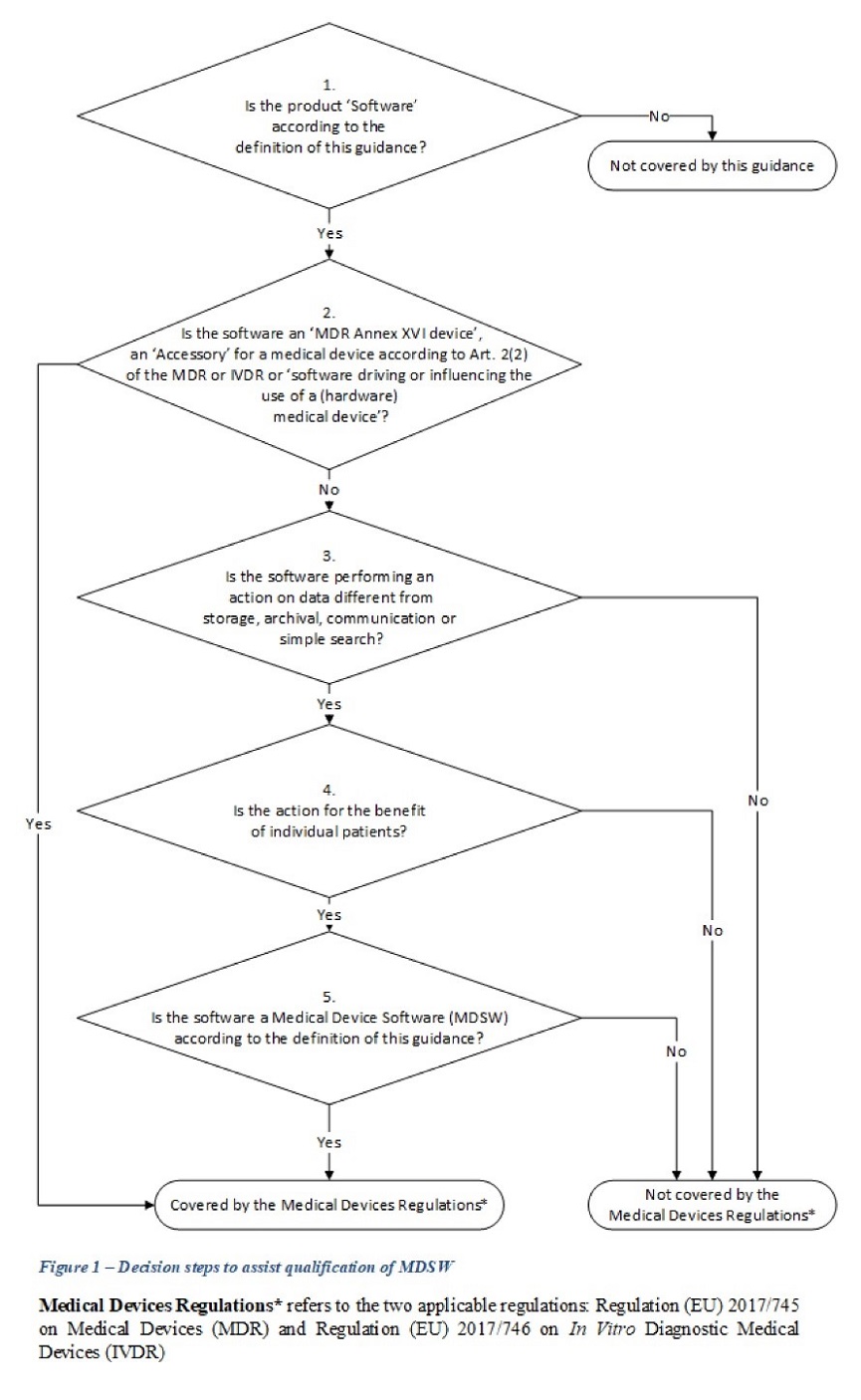
While the new guidance does not address all relevant software-specific matters, such as providing guidance on when software is considered as “placed on the market” (which is a more complicated matter for software than for hardware medical devices), it is a very welcome source of clarification that enables software manufacturers to plan accordingly. Last but not least, the guidance contains a decision tree (mirroring the one found in MEDDEV 2.1/6) to help manufacturers in the regulatory analysis of their software products.
Annex 1
Contacts

Related Resources
Capabilities
Related Pages
- Stay Up To DateSubscribe to Sidley Publications
- Follow Sidley on Social MediaSocial Media Directory